Diffusion Weighted Imaging (DWI) - Single subject
A step-by-step guide to DWI preprocessing & SC generation based on MRtrix3
This is largely adapted from Andrew Jahn’s book; consider checking the book for a thorough tutorial with MRtrix3.
CPUs
To determine the number of available logical processors or CPUs on your system, enter nproc in the terminal. Then, pass the output to the -nthreads flag below. Spare some processors if you wish to perform other tasks while the pipeline is running as it usually runs for a couple hours.
STEP 1. Preprocessing
#!/bin/bash
################################################################
# MUST FOLLOW BIDS ARCHITECTURE:
# sub
# -anat
# -*T1w.nii.gz
# -dwi
# -*.bvec
# -*.bval
# -*.json
# -*dwi.nii.gz
################################################################
############################### STEP 1 ###############################
# Convert data to .mif format and denoise #
######################################################################
# Also consider doing Gibbs denoising (using mrdegibbs). Check your diffusion data for ringing artifacts before deciding whether to use it
mrconvert *dwi.nii.gz dwi.mif
mrconvert dwi.mif -fslgrad *.bvec *.bval dwi_header.mif
dwidenoise dwi_header.mif dwi_den.mif -noise noise.mif
mrdegibbs dwi_den.mif dwi_den_unr.mif
# Extract the b0 images from the diffusion data acquired in the AP direction
dwiextract dwi_den.mif - -bzero | mrmath - mean mean_b0_AP.mif -axis 3
######################################################################
# Runs the dwipreproc command, which is a wrapper for eddy and topup.
#### !!! Here the CAMCAN dataset does not provide reverse encoding, hence -rpe_none !!!
######################################################################
dwifslpreproc dwi_den.mif dwi_den_preproc.mif -pe_dir AP -rpe_none -readout_time 0.0342002 -eddy_options " --slm=linear --data_is_shelled" -nthreads 8
# Performs bias field correction. Needs ANTs to be installed in order to use the "ants" option (use "fsl" otherwise)
dwibiascorrect ants dwi_den_preproc.mif dwi_den_preproc_unbiased.mif -bias bias.mif
########################### STEP 2 ###################################
# Basis function for each tissue type #
######################################################################
# Create a basis function from the subject's DWI data. The "dhollander" function is best used for multi-shell acquisitions; it will estimate different basis functions for each tissue type. For single-shell acquisition, use the "tournier" function instead
dwi2response dhollander dwi_den_preproc_unbiased.mif wm.txt gm.txt csf.txt -voxels voxels.mif
#Upsample the difusion image for better resolution and tracto later
mrgrid *unbiased.mif regrid -vox 1.5 dwi_unbiased_upsampled.mif
# Create a mask for future processing steps
dwi2mask dwi_unbiased_upsampled.mif mask_up.mif
# Performs multishell-multitissue constrained spherical deconvolution, using the basis functions estimated above
dwi2fod msmt_csd dwi_unbiased_upsampled.mif -mask mask_up.mif wm.txt wmfod_up.mif gm.txt gmfod_up.mif csf.txt csffod_up.mif
# Creates an image of the fiber orientation densities overlaid onto the estimated tissues (Blue=WM; Green=GM; Red=CSF)
# You should see FOD's mostly within the white matter. These can be viewed later with the command "mrview vf.mif -odf.load_sh wmfod.mif"
mrconvert -coord 3 0 wmfod_up.mif - | mrcat csffod_up.mif gmfod_up.mif - vf_up.mif
# Now normalize the FODs to enable comparison between subjects
mtnormalise wmfod_up.mif wmfod_norm_up.mif gmfod_up.mif gmfod_norm_up.mif csffod_up.mif csffod_norm_up.mif -mask mask_up.mif
########################### STEP 3 ###################################
# Create a GM/WM boundary for seed analysis #
######################################################################
# Convert the anatomical image to .mif format, and then extract all five tissue catagories (1=GM; 2=Subcortical GM; 3=WM; 4=CSF; 5=Pathological tissue)
mrconvert ../anat/*T1w.nii.gz T1.mif
5ttgen fsl T1.mif 5tt_nocoreg.mif -nthreads 8
# The following series of commands will take the average of the b0 images (which have the best contrast), convert them and the 5tt image to NIFTI format, and use it for coregistration.
dwiextract dwi_den_preproc_unbiased_upsampled.mif - -bzero | mrmath - mean mean_b0_processed_up.mif -axis 3
mrconvert mean_b0_processed_up.mif mean_b0_processed_up.nii.gz
mrconvert 5tt_nocoreg.mif 5tt_nocoreg.nii.gz
# Uses FSL commands fslroi and flirt to create a transformation matrix for regisitration between the tissue map and the b0 images
fslroi 5tt_nocoreg.nii.gz 5tt_vol0.nii.gz 0 1 #Extract the first volume of the 5tt dataset (since flirt can only use 3D images, not 4D images)
flirt -in mean_b0_processed_up.nii.gz -ref 5tt_vol0.nii.gz -interp nearestneighbour -dof 6 -omat diff2struct_fsl_up.mat
transformconvert diff2struct_fsl_up.mat mean_b0_processed_up.nii.gz 5tt_nocoreg.nii.gz flirt_import diff2struct_mrtrix_up.txt
mrtransform 5tt_nocoreg.mif -linear diff2struct_mrtrix_up.txt -inverse 5tt_coreg_up.mif
#Create a seed region along the GM/WM boundary
5tt2gmwmi 5tt_coreg_up.mif gmwmSeed_coreg_up.mif STEP 2. Streamline generation
Make sure to allocate at least 5Go of space for each subject on your local disk as streamline generation can get quite heavy. Here we generate 10 millions streamlines and then reduce that number using the tckgen and tcksift2 command.
#!/bin/bash
########################## STEP 4 ###################################
# Run the streamline analysis #
######################################################################
# Create streamlines
# Note that the "right" number of streamlines is still up for debate. Last I read from the MRtrix documentation,
# They recommend about 100 million tracks. Here I use 10 million, if only to save time. Read their papers and then make a decision
tckgen -act 5tt_coreg_up.mif -backtrack -seed_gmwmi gmwmSeed_coreg_up.mif -nthreads 8 -maxlength 250 -cutoff 0.06 -select 10000k wmfod_norm_up.mif tracks_10M_up.tck
# Extract a subset of tracks (here, 200 thousand) for ease of visualization
# tckedit tracks_10M.tck -number 200k smallerTracks_200k.tck
# Reduce the number of streamlines with tcksift
tcksift2 -act 5tt_coreg_up.mif -out_mu sift_mu_up.txt -out_coeffs sift_coeffs_up.txt -nthreads 8 tracks_10M_up.tck wmfod_norm_up.mif sift_1M_up.txt STEP 3. Recon-all
Download the lh and rh.hcpmmp1 annot files in the supplementary files here and put them into $SUBJECTS_DIR/fsaverage/label. This will prepare the T1 image for the HCP MMP 1.0 atlas (Glasser et al. 2016).
Replace
<sub>with the name of your subject.If recon-all does not work, make sure you have the tcsh shell installed by typing tcsh on the command line. In the event tcsh is not installed, enter “sudo apt install tcsh”.
If the flag -s is not recognized, use -subjid instead
#!/bin/bash
NPROC=$(nproc)
# Since the HCPMMP1-atlas is a FreeSurfer-based atlas, you have to preprocess the T1 image in FreeSurfer. This will take several hours to complete.
SUBJECTS_DIR=`pwd`;
recon-all –s <sub>_recon –i T1_raw.nii.gz –all -nthreads $NPROC
# Move into freesurfer's subjects directory. Substitute <password> for your UNIX password
echo <password> | sudo -S mv *recon $SUBJECTS_DIR
# Grant permission to write the file
echo <password> | sudo -S chmod -R a+w $FREESURFER_HOMEYou can then carry on with mapping the glasser annotation file:
################################################################
# For the command "mri_aparc2aseg", there can be an error which is due to the way multithreading is handled. Just rerun the command manually until it works or use a single thread
################################################################
# Map the annotation files of the HCP MMP 1.0 atlas from fsaverage to your subject for both hemispheres:
mri_surf2surf --srcsubject fsaverage --trgsubject <sub>_recon --hemi lh --sval-annot $SUBJECTS_DIR/fsaverage/label/lh.hcpmmp1.annot --tval
$SUBJECTS_DIR/<sub>_recon/label/lh.hcpmmp1.annot
mri_surf2surf --srcsubject fsaverage --trgsubject <sub>_recon --hemi rh --sval-annot $SUBJECTS_DIR/fsaverage/label/rh.hcpmmp1.annot --tval $SUBJECTS_DIR/<sub>_recon/label/rh.hcpmmp1.annot
mri_aparc2aseg --old-ribbon --s <sub>_recon --annot hcpmmp1 --o hcpmmp1.mgz --nthreads $NPROC/2STEP 4. Generate the Structural Connectome (SC)
#!/bin/bash
mrconvert –datatype uint32 hcpmmp1.mgz hcpmmp1.mif
# Replace the random integers of the hcpmmp1.mif file with integers
# that start at 1 and increase by 1.
labelconvert hcpmmp1.mif $MRtrix3/labelconvert/hcpmmp1_original.txt $MRtrix3/labelconvert/hcpmmp1_ordered.txt hcpmmp1_parcels_nocoreg.mif
# Register the ordered atlas-based volumetric parcellation to diffusion space.
mrtransform hcpmmp1_parcels_nocoreg.mif –linear diff2struct_mrtrix_up.txt –inverse –datatype uint32 hcpmmp1_parcels_coreg_up.mif
# Create a whole-brain connectome, representing the streamlines between each parcellation pair in the atlas (in this case, 379x379). The "symmetric" option will make the lower diagonal the same as the upper diagonal, and the "scale_invnodevol" option will scale the connectome by the inverse of the size of the node
tck2connectome -symmetric -zero_diagonal -scale_invnodevol -tck_weights_in sift_1M_up.txt tracks_10M_up.tck hcpmmp1_parcels_coreg_up.mif <sub>_hcpmmp1_parcels_coreg_up.csv -out_assignment assignments_IN_hcpmmp1_parcels_coreg_up.csv
# Visualize the connectome in MRtrix3
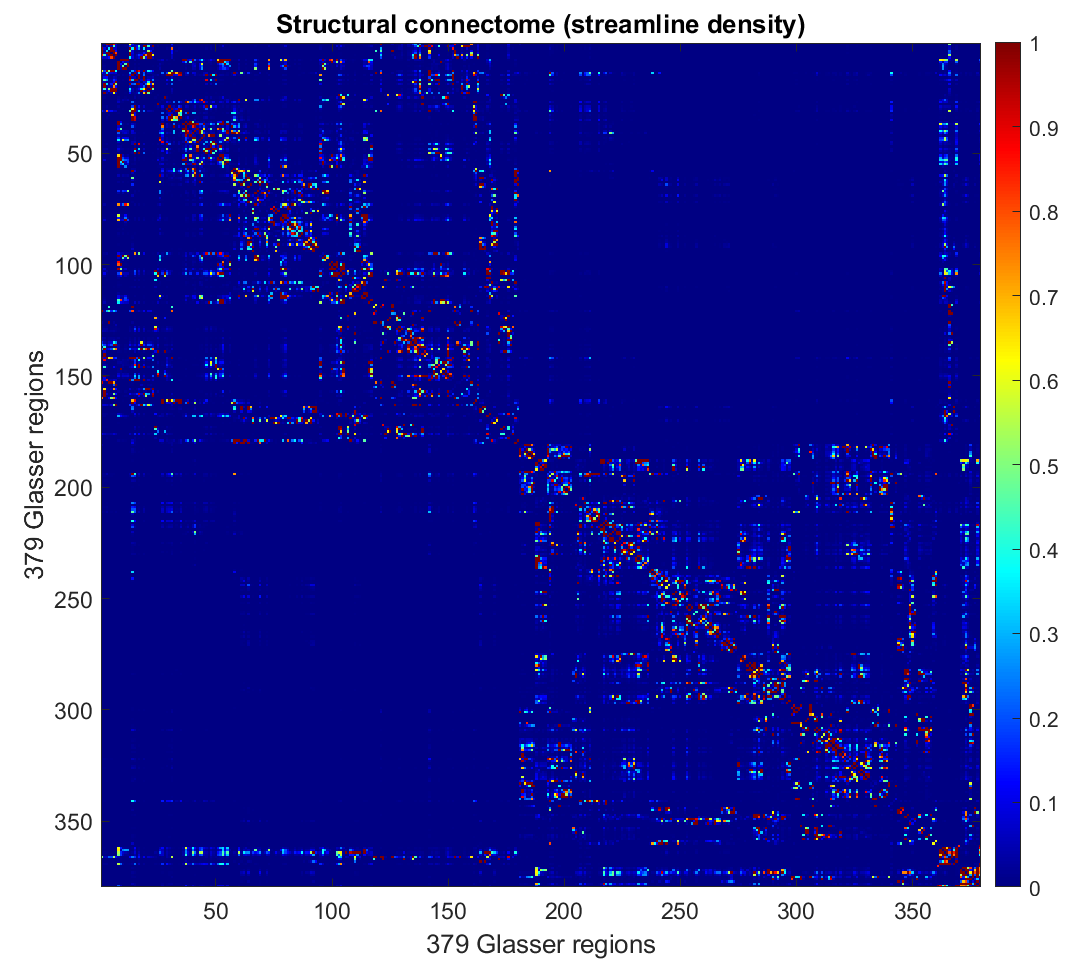
mrview hcpmmp1_parcels_coreg_up.mif -connectome.init hcpmmp1_parcels_coreg_up.mif -connectome.load sub*.csvYou can then visualize the connectivity matrix in matlab: the upper left and lower right quadrant represent the left and right intra-hemispheric connections respectively.
OPTIONAL. FA-weighted connectome
# Generate the RGB-colored FA map
dwi2tensor dwi_den_preproc_unbiased_upsampled.mif - | tensor2metric - -fa - | mrcalc - -abs FA.mif
# Generate the connectome
tcksample tracks_10M_up.tck FA.mif mean_FA_per_streamline_up.csv -stat_tck mean
tck2connectome -symmetric tracks_10M_up.tck -tck_weights_in sift_1M_up.txt hcpmmp1_parcels_coreg_up.mif mean_FA_connectome_up.csv -scale_file mean_FA_per_streamline_up.csv -stat_edge mean